
L’Autorisation de Mise sur le Marché
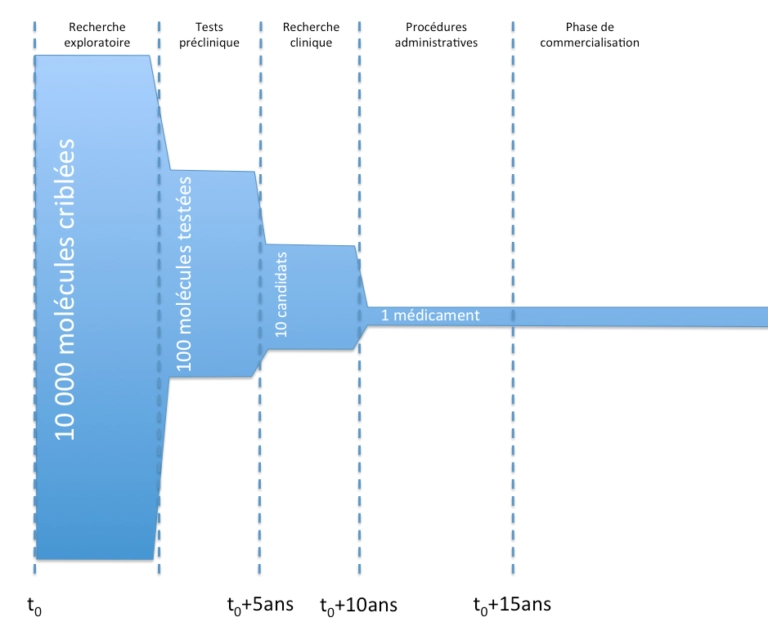
L’Autorisation de Mise sur le Marché est une procédure qui s’est généralisée dans la plupart des pays du monde, sous le contrôle des États, afin de garantir aux patients la sécurité et l’efficacité des médicaments, de leur conception à leur retrait du marché (notion de cycle de vie). Les médicaments, avant d’être introduits sur le marché européen ou français, doivent recevoir une AMM. Un médicament est légalement défini comme : Certaines revendications ou allégations thérapeutiques avancées pour certains produits cosmétiques ou compléments alimentaires peuvent donc valoir à leurs propriétaires les foudres des autorités de tutelle. L’AMM est l’aboutissement d’un long processus de conception d’un médicament sûr et efficace. Ce processus prend généralement une quinzaine d’années. D’un point de vue général, voici les principales étapes de la vie d’un médicament, de sa conception à sa commercialisation. (1) Fig.1 Vie d’un médicament et AMM (1) La recherche exploratoire combine, pour le médicament chimique, la synthèse de multiples variations à partir d’un même squelette de base, ou la réutilisation de molécules existantes dans des bases de données. Les molécules les plus susceptibles de donner une action pharmacologique sont sélectionnées soit par des tests sur des modèles in vitro, in vivo (modèles cellulaires) ou in silico (modèles informatiques, basés sur des études de docking ou QSAR[i]). À l’issue de ce « screening », seule une centaine de molécules sont éligibles pour le démarrage d’essais pharmacologiques. Finalement, pour le dépôt d’AMM, il n’en restera qu’une seule ! Viennent ensuite les essais précliniques et les essais cliniques. (1) Les essais précliniques ont pour but de vérifier la tolérance des molécules in vitro chez l’animal, de réaliser des tests toxicologiques et d’étudier la pharmacocinétique des molécules (en gros, de voir à quelle vitesse les molécules agissent, sont absorbées ou sont éliminées). La recherche clinique (environ 7 ans de travail) se décompose en trois phases principales : Pendant la phase de commercialisation, un système de « pharmacovigilance » est mis en place : les effets secondaires potentiels sont recueillis auprès des professionnels de santé et peuvent donner lieu à des études observationnelles. Par ailleurs, des études interventionnelles (dites de phase IV) peuvent être réalisées, notamment si l’AMM l’exige en raison du faible nombre de patients dans les essais cliniques. Ces études sont réalisées dans des conditions proches de la prise en charge habituelle dans le but d’identifier d'éventuels effets indésirables rares non détectés lors des phases précédentes (pharmacovigilance) et de préciser les conditions d'utilisation pour certains groupes de patients à risque. Cette phase permet d'analyser les interactions médicamenteuses et favorise la mise au point de nouvelles formes galéniques ainsi que des extensions d'indications thérapeutiques. En effet, pendant la phase de commercialisation, des études cliniques secondaires peuvent être menées à l’initiative du fabricant afin de tester son médicament sur une autre pathologie, une nouvelle population, dans le cadre d’une nouvelle stratégie thérapeutique, etc. afin d’obtenir l’approbation d’une nouvelle indication. [i] Quantitative Structure-Activity Relationships Une fois obtenu le précieux sésame, il faut continuer à pouvoir protéger le médicament. Pour ce faire, diverses dispositions existent. Les médicaments sont brevetables en vertu du droit commun des brevets. Les brevets pharmaceutiques sont délivrés, comme tous les autres brevets, pour une durée de 20 ans à compter du dépôt et moyennant le paiement d’annuités. Cependant, les règles de durée concernant le brevet de médicament diffèrent du régime général du droit des brevets. En effet, les médicaments nécessitent une autorisation de mise sur le marché (AMM) avant de pouvoir être commercialisés. Cette autorisation n'est généralement pas délivrée avant plusieurs années. Le médicament ne serait donc en réalité protégé par le brevet que pendant une dizaine d’années. Aussi, pour compenser cette période durant laquelle le brevet ne peut être exploité, un titre spécial a été créé, le Certificat Complémentaire de Protection (CCP), qui prolonge les droits du titulaire d’un brevet sur un produit pharmaceutique pour une durée de 5 ans, ce qui compense le problème de l’attente de l’AMM qui peut prendre plusieurs années et pendant laquelle le médicament ne peut pas être commercialisé. La demande de certificat complémentaire de protection communautaire doit être déposée dans les six mois suivant l’obtention de l’autorisation de mise sur le marché ou dans les six mois suivant l’obtention du brevet si le brevet a été délivré après l’autorisation de mise sur le marché. Le certificat complémentaire de protection communautaire est accessoire au brevet en ce sens que si le brevet de médicament est invalide, le certificat l’est également. Il est possible de breveter une molécule déjà connue et pour laquelle une première application a déjà été divulguée si la demande de brevet porte sur une nouvelle application thérapeutique de cette molécule. Depuis le 1er juillet 2019, le règlement (CE) n° 469/2009 concernant le certificat complémentaire de protection pour les médicaments a été modifié afin de permettre aux fabricants de génériques et de biosimilaires établis dans l'Union de fabriquer dans l'Union des produits ou des médicaments contenant ces produits, à des fins d’exportation vers les marchés de pays tiers ou de stockage en attendant l’expiration d’un certificat. Cette modification introduit une exception limitant la protection conférée par un certificat et prévoit que certains actes qui nécessiteraient autrement le consentement du titulaire du certificat peuvent être réalisés sans être considérés comme une atteinte aux droits du titulaire… Concrètement, le médicament est en moyenne protégé commercialement pendant une quinzaine d’années (durée de validité du brevet au moment de la commercialisation prolongée du CCP). Finalement, la durée de protection accordée par le CCP est de : Fig.2 Durée de protection conférée par un CCP (Règlement UE) (1) Prolongation pédiatrique Si le titulaire d’un CCP fournit les résultats des études réalisées selon un plan d’investigation pédiatrique (en partie ou en totalité sur la tranche d’âge 0 à 18 ans) approuvé dans son dossier d’AMM ou d’AMM complémentaire, ce dernier a droit à une prolongation de 6 mois de son CCP. Toutefois, cette prolongation n’est pas possible : Bibliographie, pour plus d’informations :L'Autorisation de Mise sur le Marché : une garantie de sécurité et d'efficacité pour les patients
Principales étapes de la conception d’un médicament avant l’Autorisation de Mise sur le Marché
BREVET et CCP
